Disposición 5640/2022
Presento al lector la actualización (al año 2022/julio) de la exposición dinámica (revisada, concordada y comentada) de las normas técnico-jurídicas que regulan la bioequivalencia y biodisponibilidad, con fines de autorización de medicamentos, según la Ley 16.463.
Elaboración propia siguiendo una metodología que me pertenece (y que he aplicado en la obra «Ley 16.463 Comentada y Concordada» y en 2017 en el proyecto de Digesto de normas federales de medicamentos). Metodología consistente con los criterios jurídicos del Digesto Jurídico Argentino al que le he sumado la consolidación de los textos; se revisan las fuentes oficiales primarias con la dinámica del Derecho regulatorio de los productos para la salud. La versión completa cuenta con los links de los textos normativos actualizados (revisados y vigentes).

BIOEQUIVALENCIA. PROCEDIMIENTO ACTUAL
Disposición 5640/2022
La solicitud de autorización de estudios de bioequivalencia in vivo que se realicen en el país así como la presentación de los resultados se regirán por el procedimiento establecido en la presente disposición.
Texto de la norma
BIOEXENCIONES
Anexo I Disposición 5068-2019 (Vigente): GUÍA PARA LA SOLICITUD DE BIOEXENCIONES DE IFA CON REQUERIMIENTO DE BIOEQUIVALENCIA; Anexo II y III: solicitud y documentación obligatoria. (Abroga la Disposición ANMAT Nº 6766/16).
Abroga la Disposición ANMAT N° 6766/16. Relacionada con la Disposición ANMAT Nº 271/19 (abrogada) Programa Integral de Biodisponibilidad, Bioequivalencia e Intercambiabilidad de Medicamentos y la Disposición ANMAT N° 758/09.
Bioequivalencia y Biodisponibilidad
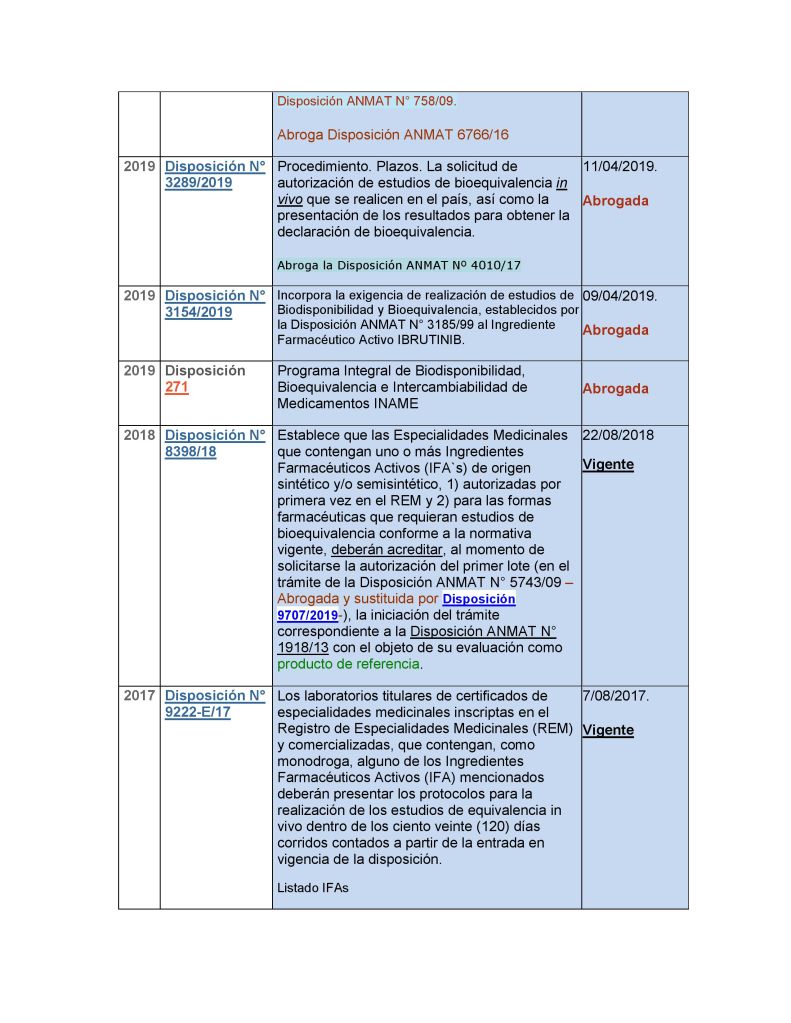
Disposición N° 8398/18 (vigente)
ARTÍCULO 1º.- Establécese que las Especialidades Medicinales que contengan uno o más Ingredientes Farmacéuticos Activos (IFA`s) de origen sintético y/o semisintético, autorizadas por primera vez en el REM y para las formas farmacéuticas que requieran estudios de bioequivalencia conforme a la normativa vigente, deberán acreditar, al momento de solicitarse la autorización del primer lote en el marco de la Disposición ANMAT N° 5743/09 (abrogada y sustituida), la iniciación del trámite correspondiente a la Disposición ANMAT N° 1918/13 a los fines de su evaluación como producto de referencia.
ARTÍCULO 2º.- Establécese que en las solicitudes de inscripción en el REM de medicamentos multifuente de productos que correspondan a IFAs autorizados por primera vez en el mercado argentino, y con producto de referencia declarado conforme a la normativa aplicable, deberá incluirse la acreditación del inicio del trámite correspondiente a la autorización del protocolo del estudio de equivalencia in vivo o in vitro, según los requerimientos de las disposiciones vigentes.
ARTICULO 3º.- Establécese para los productos referidos en el artículo 2º, que cuando se presenten solicitudes de autorización del primer lote en el marco de la Disposición ANMAT N° 5743/09, se deberá presentar la disposición emitida por esta Administración Nacional que los declare equivalentes al producto de referencia, previamente establecido de acuerdo al artículo 1° de la presente disposición, en base a los estudios de equivalencia in vivo o in vitro realizados.
Disposición 9222/17 (Vigente).
Los laboratorios titulares de certificados de especialidades medicinales inscriptas en el Registro de Especialidades Medicinales (REM) y comercializadas, que contengan, como monodroga, alguno de los Ingredientes Farmacéuticos Activos (IFA) mencionados a continuación, deberán presentar los protocolos para la realización de los estudios de equivalencia in vivo dentro de los ciento veinte (120) días corridos contados a partir de la entrada en vigencia de la presente disposición. (ver disposición DI-2017-9222-APN-ANMAT#MS del 3/8/17 BO 7/8/17 Disposicion_9222-E-2017).
Los Ingredientes Farmacéuticos Activos (IFA) referidos precedentemente son los siguientes:
-Acenocumarol -Clopidogrel -Clozapina -Haloperidol -Levotiroxina
-Nitrofurantoina -Olanzapina -Quetiapina -Risperidona.
Disposición 8870/17.
Las especialidades medicinales que contengan los Ingredientes Farmacéuticos Activos (IFAs), como monodroga, indicados en el Anexo I de la presente, y para todas sus concentraciones autorizadas, no requerirán la realización de estudios de bioequivalencia (ver disposición 8870/2017 ANMAT (Vigente). BO 2-8-2017 Disposicion_8870-2017).
INGREDIENTES FARMACÉUTICOS ACTIVOS QUE NO REQUIEREN DEMOSTRACIÓN DE BIOEQUIVALENCIA
1.- ACARBOSA
2.- BISACODILO
3.- COLESTIRAMINA
4.- DIMETICONA (SIMETICONA)
5.- HOMATROPINA (Empleo farmacológico)
6.- LACTULOSA
7.- MEBENDAZOL
8.- NISTATINA
9.- ORLISTAT
10.- PICOSULFATO SÓDICO
11.- POLICARBOFILO SÓDICO
12.- POLICARBOFILO CÁLCICO
13.- SUCRALFATO.
Antecedentes técnicos:
Marco para la Ejecución de los Requisitos de Equivalencia para los Productos Farmacéuticos
Red PARF Documento Técnico Nº 8 Red Panamericana de Armonización de la Reglamentación Farmacéutica OPS 2011 (Ver el documento: Red PARF Documento Técnico Nº 8 )
Antecedentes normativos:
Disposición 3185/99:
Apruébanse las recomendaciones técnicas para la realización de estudios de equivalencia contenidas en el documento: «Cronograma para exigencia de estudios de equivalencia entre medicamentos de riesgo sanitario significativo» cuyo texto se reproduce como Anexo I de la presente Disposición formando parte de la misma. Listado de IFAS clasificadas por CATEGORÍAS DE RIESGO SANITARIO
Disposiciones relacionadas:
Adóptanse los Criterios de Bioexención de Estudios de Bioequivalencia, basados en la Clasificación Biofarmacéutica para las formas farmacéuticas sólidas orales de liberación inmediata, con alcance para algunos de los principios activos establecidos en la Disposición (ANMAT) Nº 3185/99 como de riesgo intermedio y los que en un futuro se vayan incorporando, así como las exigencias que se deberán cumplir para demostración de la Bioexención, los cuales figuran como Anexo I de la presente Disposición.
Disposición Nº 1263/2012
Realización del estudio de bioequivalencia o equivalencia “in vitro”, según corresponda, el laboratorio patrocinante deberá proponer alguna de las tres opciones: a) tres lotes vigentes, de escala industrial, idénticos al producto a comercializarse, b) tres lotes pilotos de tamaño no menor a las 100.000 unidades o c) tres lotes pilotos sin establecer un tamaño mínimo de unidades.
Disposición 4010/2017 (abrogada)
ARTÍCULO 1°.- Déjanse sin efecto los Anexos II, III y IV de la Disposición ANMAT N° 5040/06.
ARTÍCULO 2°.- Apruébase el nuevo formulario para la solicitud de autorización para la realización de estudios de biodisponibilidad/bioequivalencia con las pautas y requisitos de información y documentación que figura como Anexo I de la presente disposición y forma parte integrante de la misma.
ARTÍCULO 3°- Apruébase el nuevo formulario para la presentación de resultados de estudios de biodisponibilidad/bioequivalencia con las pautas y requisitos de información y documentación, que figura como Anexo II de la presente Disposición, que forma parte integrante de la misma.
En aplicación: RÉGIMEN DE BUENAS PRACTICAS PARA LA REALIZACIÓN DE ESTUDIOS DE BIODISPONIBILIDAD / BIOEQUIVALENCIA (Anexo I Disposición ANMAT N° 5040/06 (actualizada 2017)
Antecedentes de Disposición ANMAT N° 5040/06:
Disposición ANMAT N° 5040/06 (actualizada 2017)
modificada por Disposición N° 1746/2007 y Disposición N° 1263/2012 y Disposición N° 4010/2017
RÉGIMEN DE BUENAS PRACTICAS PARA LA REALIZACIÓN DE ESTUDIOS DE BIODISPONIBILIDAD /BIOEQUIVALENCIA (Anexo I, sustituido por art. 1° de la Disposición N° 1746/2007 ) y pautas y requisitos de información y documentación para la solicitud de autorización para la realización de estudios BE/BD (Anexo II ELIMINADO), requisitos de información y documentación DE LA ETAPA BIOANALÍTICA (Anexo III ELIMINADO), forma de presentación de los resultados de los estudios de biodisponibilidad/bioequivalencia (Anexo IV ELIMINADO).
Circular Administrador 11 de 2016
Disposición 6766/2016 (abrogada) Guía para la solicitud de Bioexenciones de Ingredientes Farmacéuticos Activos con Requerimiento de Bioequivalencia
Tabla dinámica del estado de las normas de BD y BE y sus relaciones, revisadas, comentadas, concordadas, tomadas de fuentes oficiales: